МАТЕРИАЛЫ ПО ЭКОЛОГИИ
Гигиенические критерии состояния окружающей среды для полихлорированных бифенилов и терфенилов
22 мая 2009 г.
2. СВОЙСТВА И АНАЛИТИЧЕСКИЕ МЕТОДЫ
2.1 Химический состав
ПХБФ составляют целый класс хлорированных углеводородов и производятся в коммерческих целях путем прогрессирующего хлорирования бифенила в присутствии подходящего катализатора. Они известны под различными торговыми названиями: Арохлор (США), Фенохлор (Франция), Хлофен (Федеративная Республика Германии), Канехлор (Япония), Фенхлор (Италия) и Совол (СССР). Их значимость для промышленного применения зависит от химической инертности, устойчивости к нагреванию, негорючести, низкому давлению паров (особенно у высокохлорированных соединений) и высокой диэлектрической постоянной. Имеется много различных торговых названий смесей ПХБФ с другими соединениями.
Отдельные производители имеют собственные системы идентификации выпускаемых продуктов. В серии "Арохлор" используется четырехзначный код. Бифенилы обычно обозначаются цифрой 12, занимающей первые два места, в то время как последние две цифры обозначают процент по массе содержания хлора в смеси; таким образом, Арохлор 1260 представляет собой смесь полихлорированных бифенилов, содержащих 60% хлора. Исключением из этого общего правила является Арохлор 1016, который является продуктом дистилляции Арохлора 1242, содержащим только 1% компонентов; с 5 или большим числом атомов хлора (Burse et al., 1974). В других коммерческих продуктах коды могут обозначать примерное среднее число атомов хлора в составляющих компонентах. Так, Хлорфен А60, Фенохлор ДР6 и Канехлор 600 являются бифенилами со средним числом 6 атомов хлора в молекуле (эквивалент 59% содержания хлора по массе).
В серии "Арохлор" терфенилы обозначаются цифрой 54, занимающей первые два места четырехзначного кода. В Японии ПХТФ закодированы под названием Канехлор КС-С.
Отдельные ПХБФ были синтезированы для использования в качестве эталонных проб, применяемых при идентификации газо-жидкостных хроматографических пиков, при проведении токсикологических исследований и изучении их метаболических превращений в живых организмах. Для этих целей эти соединения готовятся меченными 14С (Hutzinger et al., 1971; Tas и de Vos, 1971; Webb и McCall, 1972; Moron et al., 1972; Sundstrom и Wachtmeister, 1973; Jensen и Sundstrom, 1974).
Хлорирование бифенила может привести к замещению от 1 до 10 атомов водорода хлором. Обычный порядок положений такого замещения показан на нижеприведенном рисунке.. Теоретически возможно получить 210 различных бифенилов с различным содержанием хлора, хотя Sissons и Welti (1971) показали, что замещение хлора в положениях 3,5 и 2, 4, 6 при прямом хлорировании бифенила не происходит. Содержание ПХБФ в Арохлорах с замещением хлором 1-9 атомов водорода отражено в табл. 1.
Таблица 1. Приблизительный состав Арохлоров

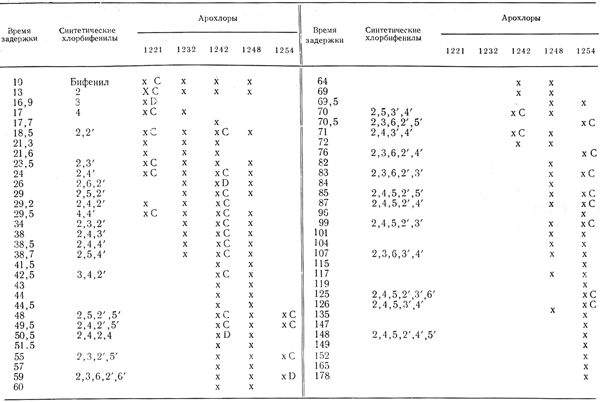
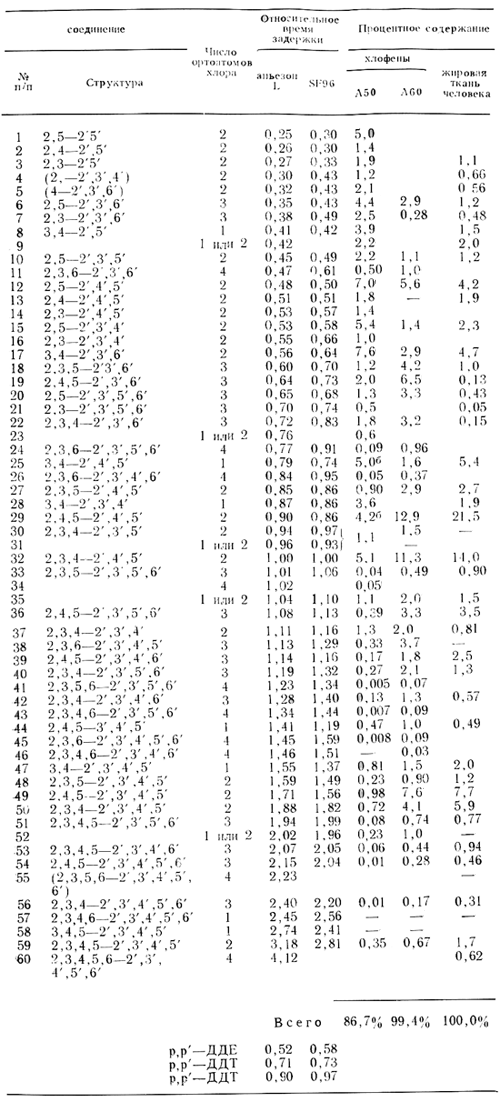
Были проведены многочисленные исследования с целью идентификации отдельных ПХБФ в коммерческих продуктах. Sissons и Welti (1971) разделили компоненты Арохлоров с помощью колонковой и газо-жидкостной хроматографии и охарактеризовали многие пики с помощью масс-спектромет-рии с высокой степенью разрешения и методом ядерного магнитного резонанса, а также путем сравнения с 40 синтезированными ПХБФ. Webb и McCall (1972) идентифицировали газо-жидкостные хроматографические пики с таковыми, характерные для синтезированных соединений, путем определения времени задержки и инфракрасной спектрометрии (табл. 2). Наиболее исчерпывающее исследование было выполнено Jensen и Sundstrom (1974). Ими было показано, что с помощью обычной газо-жидкостной хроматографии разделить все компоненты невозможно, поэтому они проводили предварительное фракционирование на колонке с активированным углем, которая позволяет разделять компоненты ПХБФ в соответствии с нахождением замещающих атомов хлора в молекуле в положениях 2, 2', 6 или 6' (атомы орто-хлора). Они сравнили пики, получаемые при осуществлении газо-жидкостной хроматографии, с таковыми, характерными для 90 синтезированных ПХБФ, и получали возможность охарактеризовать в том числе и количественно 60 компонентов Хлофена А50 и А60 (табл. 3). Данные табл. 2 и 3 свидетельствуют о значительном совпадении компонентов Арохлора 1254 и Хлофена А50.
Таблица 2. Полихлорбифенилы в Арохлорах 1221-1254 (Webb и McCall, 1972)
2.2 Чистота продуктов
Коммерческие ПХБФ продают без указания их состава, но со спецификацией физических свойств. Разные партии ПХБФ могут несколько отличаться от составов, указанных в табл. 1-3. Примеси, содержащиеся в коммерческих ПХБФ, являются хлорированными дибензофуранами и нафталинами (Vos et al., 1970; Bowes et al., 1975). Bowes и др. (1975) обнаружили хлорированные дибензофураны в концентрации 0,8-40 мг/кг в образцах Арохлоров 1248-1260 (не обнаружив их в Арохлоре 1016), 8,4 мг/кг в Хлофене А60 и 13,6 мг/кг в ч>енохлоре ДР-6. Roach и Pomerantz (1974) обнаружили хлорированные дибензофураны на уровне 1 мг/кг, a Nagayama и др. (1976) - 18 мг/кг в различных партиях Кане-хлора 400, не обнаружив в них хлорированных дибензодиоксинов.
2.3 Определение остатков ПХБФ
Были опубликованы обзоры о методах, используемых для определения хлорорганических соединений, включая ПХБФ в пробах, отобранных из окружающей среды (Holden, 1973a, Panel on Hazardous Trace Substances, 1972). Нельзя найти двух лабораторий, использующих совершенно идентичные методы определений, хотя последние имеют много общих черт. Применяемая техника представляется той же, которая была ранее разработана для определения хлорорганических пестицидов с соответствующими изменениями, связанными с присутствием ПХБФ; исследования ПХБФ зачастую составляют часть более широкой программы постоянного надзора за стойкими хлорорганическими соединениями в окружающей среде. Наибольшую трудность в определении ПХБФ составляет необходимость отделения их от мешающих определению хлорорганических пестицидов и в получении однозначной количественной характеристики широко варьирующих смесей компонентов.
2.3.1 Извлечение из образца
Воздух
Взвешенные частицы, попадающие из воздуха, улавливались нейлоновой сеткой с величиной пор 200 мкм, покрытой силиконовой смазкой с последующим экстрагированием ПХБФ гексаном (Sodergren, 1972a). Были проделаны раздельные определения фаз ПХБФ, содержащихся в виде взвешенных частиц и паров в воздухе, путем пропускания больших объемов воздуха через фильтр, вслед за которым устанавливалась ловушка, содержащая гексан (Hasegawa et al., 1972b), полиуретановая ячейка (Bidleman и Olney, 1974) или керамическое осаждающее устройство, покрытое силиконовой смазкой марки OV17 (Harvey и Steinhauer, 1974) с целью поглощения проходящих паров.
Таблица 3. Процентное содержание полихлорированных бифенилов в Хлофенах А50, А60 и в жировой ткани человека (Jensen Sundstrom, 1974)
Вода
Полихлорбифенилы экстрагируются из воды путем пропускания пробы через фильтр из ундекана и моностеарата Кар-бовакса-400 с подложкой из хромосорба W (Ahling и Jensen, 1970) или через пористую ячейку из полиуретана, покрытую соответствующей газо-жидкостной хроматографи-ческой стационарной "фазой (Uthe et al., 1972) или амберлито-вой смолой ХАД-2 (Harvey et al., 1973), с последующим вымыванием ПХБФ растворителем. Anhoff и Josefsson (1975) описали жидкостно-жидкостную экстракцию с помощью цик-логексана.
Биологические образцы
Большинство аналитиков использует стандартные методы, разработанные для хлорорганических пестицидов, при использовании которых ПХБФ экстрагируется вместе с жирами, образец фиксируется с помощью безводного сульфата натрия и затем экстрагируется петролейным эфиром или гек-саном. Porter и др. (1970) изучили оптимальные условия осуществления этой процедуры. Для облегчения расщепления клеточных структур может быть включен обезвоженный растворитель; также использовали этанол (Noren и Westoo, 1968) и ацетон (Jensen et al., 1973). Rote и Murphy (1971) обрабатывали образцы смесью уксусной и перхлорированной кислот до экстракции гексаном.
2.3.2 Очистка
Методы удаления жира из экстракта включают в себя разделение гексаном и ацетонитрилом или диметилформамидом, обработку крепкой серной кислотой или спиртовым раствором гидроокиси калия. Также используется метод проницаемости геля (Stalling et al., 1972); Holden и Marsden (1969) применили метод осаждения жира на сухой частично деактивированной алюминиевой колонке. Некоторые пестициды, такие как диелдрин, разрушаются серной кислотой, поэтому этот метод применять не следует, если подобные пестициды необходимо определять вместе с ПХБФ (Jensen et al., 1973).
ПХБФ могут быть отделены от хлорорганических пестицидов с помощью колонковой хроматографии на флоризиде (Mulhern et al., 1971), силикагеля (Holden и Marsden, 1969; Armour и Burke, 1970; Collins et al., 1972) или на колонке с активированным углем (Berg et al., 1972; Jensen и Sundstrom, 1974). В ряде лабораторий отмечали трудности воспроизведения результатов, получаемых другими исследователями. Легкость разделения образца оказалась зависимой от характеристик абсорбента, от типа элюирующего растворителя, от самого экстрагируемого образца при отсутствии больших трудностей в отделении любых интерферирующих субстанций, за исключением ДДЕ, являющегося метаболитом ДДТ. Метод тонкослойной хроматографии использовали Noren и Wes-too (1968), Nagley и др. (1970) и Reinke и др. (1973). Во многих образцах, отобранных из окружающей среды, ДДЕ присутствует в больших количествах по сравнению с ПХБФ и должен быть удален до начала количественного определения ПХБФ. Для превращения ДДЕ в дихлорбензофе-нон использовали процессы окисления; в качестве окислителей применяли бихромат калия и серную кислоту (Westoo и Noren, 1970) и окись хрома (II) и уксусную кислоту (Mulhern et al., 1971). Jensen и Sundstrom (1974), определявшие соотношения ДДТ/ПХБФ в образцах, отобранных из окружающей среды, предпочли использовать раствор бихромата натрия в уксусной кислоте с добавлением следовых количеств серной кислоты. Они заявили, что применение данного способа не разрушает ДДТ и его метаболит ДДЕ, которые могут присутствовать в экстрактах после обработки образцов крепкой серной кислотой, и что применение этой смеси делает возможным количественное определение дихлорбензофенона, получаемого при окислении ДДЕ.
Превращение ДДТ в ДДЕ может быть достигнуто путем обработки спиртовым раствором гидроокиси калия, который также устраняет интерференцию, вызванную присутствием элементарной серы (Ahling и Jensen, 1970). Сера может быть также удалена активированным скелетным никелевым катализатором гидрирования (Ahnoff и Josefsson, 1975) или металлической ртутью.
Sodergren (1973b) сумел добиться разделения малых образцов путем использования объемов порядка микролитров.
2.3.3 Хроматографическое разделение ПХБФ
Газо-жидкостная хроматография
Большинство аналитиков использует газо-жидкостную хроматографию с детектором в виде электронной ловушки для отделения ПХБФ от экстракта после разделения образца. В качестве стационарной фазы обычно используются силиконы или их производные, например DC200, SF96, OV1, QF1 или Апьезон L. Jensen и Sundstrom (1974) установили, что при использовании смеси из SF/96 и QF1 у Хлофена А50 можно определить 14 пиков. При применении Апьезона L можно получить еще лучшее разрешение. Эти авторы получили лучшее разделение пиков за счет предварительного фракционирования на колонке с активированным углем, которая разделяла ПХБФ в зависимости от числа ортохлорных заместителей; они считают такую очистку необходимой при анализе остаточных количеств ПХБФ, хотя такая процедура может оказаться ценной при изучении селективной деградации ПХБФ в окружающей среде. Температуры колонок варьировали в пределах 170-230°С. Колонки со стеклянными капиллярами позволяли получать хорошее отделение ПХБФ от ДДТ и его метаболитов (Schulte и Acker, 1974).
Тонкослойная хроматография
Этот метод использовали при разделении образцов, но он также обеспечивает полуколичественную оценку результатов путем визуальной оценки пятен с последующей денеитомет-рией или путем сравнения с пятнами, образуемыми известными количествами ПХБФ. Mulhern и др. (1971) разделяли ПХБФ на пластинах, покрытых алюминием, содержащим нитрат серебра, с последующим проявлением пятен в ультрафиолетовом свете; предел чувствительности различения при этом составил 1 мкг. Collins и др. (1972) разработали аналогичный метод, в котором ПХБФ группировались вместе в едином пятне; достигнутый предел разрешения при этом составил 50 нг. Метод фазово-контрастной хроматографии на пластинах, покрытых кизельгуром и обработанных жидким парафином, использовали для разгона фенохлора ДР-6 в несколько пятен с пределом различения в несколько микрограммов (de Vos и Peet, 1971).
2.3.4 Количественное определение содержания ПХБФ
Эффективность электронной ловушки неодинакова для всех компонентов ПХБФ; на ее величину оказывает большое влияние степень хлорирования (Zitko et al., 1971). Это не вызывает трудностей, когда исследуемый образец непосредственно контаминирован коммерческой смесью ПХБФ, поскольку такая смесь может использоваться в качестве эталона. Трудности появляются, когда ПХБФ в образце подвергаются селективной деградации под влиянием окружающей среды (см. разделы 3 и 5). Несколько исследователей подметили, что наблюдаемые пики при исследовании таких образцов достаточно близко совпадают с картиной, получаемой от более вы-сокохлорированных смесей ПХБФ, таких как Арохлор 1254, и что им приходилось сравнивать общую область пиков с таковой, характерной для наиболее близкого коммерческого продукта при определении содержания ПХБФ в образце (Armour и Burke, 1970). Collins и др. (1972) наблюдали, что в их условиях области пиков, которые обычно встречались при анализе экстрактов из тканевых проб, близко напоминали таковые, характерные для эквивалентных количеств ДДЕ, который мог быть использован для тарирования измерений. Для более уверенного применения такой процедуры Rote и Murphy (1971) разделили пики на группы в соответствии с числом атомов хлора в молекуле согласно масс-спектрографическим данным и подсчитали содержание ПХБФ в каждой группе, исходя из теоретически рассчитанного ответа детектора на определенное содержание хлора. Jensen и др. (1973) отобрали такой коммерческий образец ПХБФ, который включал все пики исследуемого экстракта; они определяли содержание ПХБФ в каждом пике с помощью комбинации масс-спектрометрии и кулонометрии с последующим определением общего содержания ПХБФ в образце путем сравнения высоты каждого пика, полученного при исследовании экстракта, с таким же пиком, получаемым от образца, используемого в качестве эталонного. Более простые методы использовали Коегпап и др. (1969), которые сравнивали высоту единичного пика, получаемого при исследовании экстракта, с подобным пиком, который имел то же самое время задержки, полученным при исследовании коммерческой смеси ПХБФ; другие авторы усредняли для подобного расчета данные, характерные более чем для одного пика (Rejnolds, 1971; Reinke et al., 1973). Rote и Murphy (1971) подсчитали, что подобная процедура может более чем удвоить значения, получаемые с помощью более точных методов.
Иная техника, заключающаяся в том, что ПХБФ хлорируется до насыщения с помощью пентахлорида сурьмы и превращается в декахлородифенил, который затем может быть определен с помощью одного пика, была рекомендована Berg и др. (1972).
2.3.5 Точность определений ПХБФ
Группа, состоящая из 8 аналитиков, занимающаяся изучением загрязнения Северного моря, провела совместное исследование по определению содержания ПХБФ в образце рыбьего жира с помощью методов, которые в настоящее время применяются в их лабораториях (International Council for the Exploration of the Sea, 1974). Полученные величины концентрации ПХБФ варьировали от 1,0 до 3,9 мг/кг при среднем значении 1,97 мг/кг и стандартном отклонении 0,93 мг/кг. Лучшее совпадение результатов было получено при исследовании одного и того же образца рыбьего жира с искусственно повышенной концентрацией ПХБФ порядка 10 мг/кг; средняя величина результатов определений содержания ПХБФ составила 10,0 мг/кг со стандартным отклонением 1,1 мг/кг.
Вероятным источником ошибок в данном случае является неполное первоначальное извлечение ПХБФ из образца (Holden и Marsden, 1969). Другой источник различий между данными различных лабораторий скрыт в самом методе, используемом для количественной характеристики пиков газожидкостных хроматограмм (раздел 2.3.4); Van Hove Holdri-net (1975) считает, что этот элемент является основным источником ошибок.
Совершенно очевидно, что при интерпретации аналитических результатов, получаемых в лаборатории, надо проявлять осторожность, в частности в отношении образцов с низким содержанием ПХБФ, в особенности, если компетенция лаборатории не была подтверждена путем проведения совместных межлабораторных исследований.
2.3.6 Подтверждение идентичности
После того как Jensen впервые показал с помощью масс-спектрографа, что субстанциями, создававшими помехи при газо-жидкостной хроматографии хлорорганических пестицидов, являются ПХБФ, другие исследователи также получили подтверждение присутствия ПХБФ в образцах, отобранных из окружающей среды путем комбинирования методов газо-жпдкостной хроматографии и масс-спектрометрии (Bagley et al., 1970), а также с помощью кулонометрии, которая позволяет определять содержание хлора. Превращение ПХБФ в бициклогексил и декахлорбифенпл является дальнейшим подтверждением того же положения (Berg et al.,. 1972). Широкое распространение ПХБФ в окружающей среде в настоящее время хорошо установлено, и поскольку стали доступными методы устранения помех, создаваемых присутствием хлорорганических пестицидов, исчезли доказательства присутствия других веществ, могущих создавать помехи в тех типах образцов, которые подвергались анализу вплоть да нижнего предела определения порядка 0,01 мг/кг. Это утверждение не всегда применимо к другим типам образцов, особенно при подозрениях о наличии очень низких уровней ПХБФ; Ahnoff и Josefsson (1973, 1975) сообщили о ряде неизвестных веществ, служащих помехой при определении ПХБФ з воде с содержанием их менее 1 мг/л, одно из которых в последующем было идентифицировано как элементарная сера. Для таких образцов они рекомендовали делать подтверждение с помощью масс-фрагментографии.
2.4 Определение ПХТФ
Для определения ПХТФ используют несколько методов. Процедуры извлечения и разделения здесь те же, что и при определении ПХБФ, но детали проведения газо-жидкостной хроматографии различны вследствие более низкой летучести ПХТФ. Zitko и др. (1972b) использовали в качестве стационарной фазы в колонке с температурой 200°С 3% OV210. Thomas и Reynolds (1973) также использовали OV 210 в колонке с температурой 250°С и другую систему с 3% дексилом в качестве стационарной фазы при 300°С с детектором в виде 63 Ni - электронной ловушки; ту же методику использовали Addison и др. (1972). Sosa-Lucero и др. (1973) использовали OV 210 и SE 30 при 255°С, в то время как Frendental и Greve (1973) использовали OV 17 при температуре, изменявшейся по программе от 200 до 285°С. Thomas и Reynolds (1973) подтвердили идентичность указанных соединений путем хлорирования их пентахлоридом сурьмы до тетрадекахлортер-фенила.
Также была описана техника тонкослойной хроматографии с пределом определения порядка 1 мкг (Addison et al., 1972).